ARCH_Pred server
Information
ARCH_Pred is a web application that interface a knowledge-based loop structure prediction method described in this publication. Given a query loop of unknown structure, ARCH_Pred identifies the most suitable loops from a library of structures of protein loops. The prediction algorithm shown below include 3 major steps: Selection, Filtering and Ranking.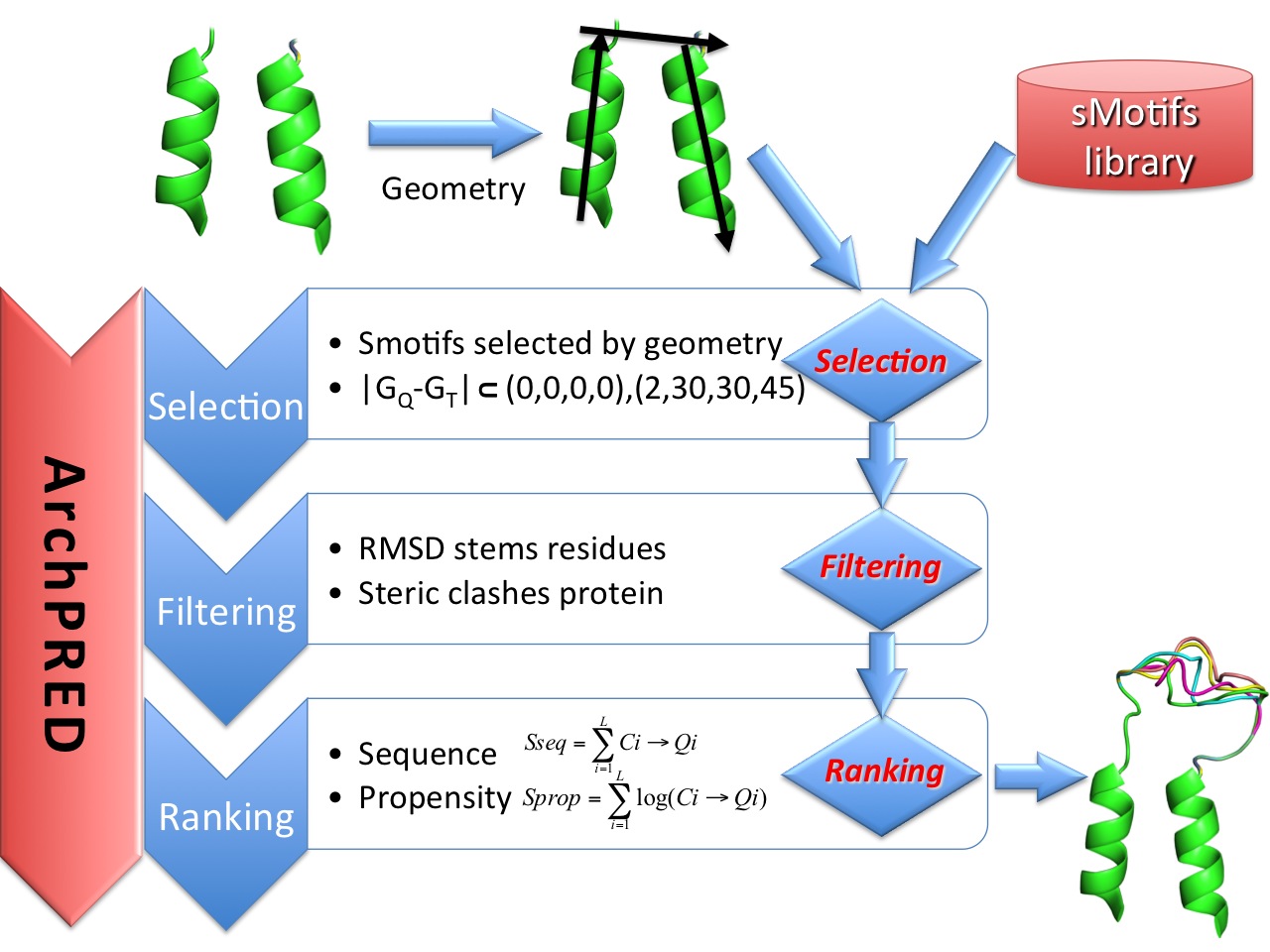
Selection
In this step, the geometry of the unknown loop region is used to query a large database of sMotifs extracted from protein structures. Smotifs will qualify if the the different in geometry between query and target falls within the semi-open interval [(0,0,0,0)-(2,30,30,45)], i.e. two loops: loop A and B with geometry GA=(DA,δA,θA, σA and GB=(DB,δB,θB,σB) share the same geometry if the difference in geometry falls inside the semi-open interval [(0,0,0,0),(2,30,30,45)).Two loopsFiltering
The smotifs selected in the previous steps undergo a filtering step based on two criteria: (i) RMSD of stem residues, i.e. residues that flank missing loop (ii) Steric clases with protein The filtering step in the algorithm discards clearly unfavorable candidates by assessing the fit of stem regions and by steric fitting in the new protein framework. The terms of steric violations or clashes are computed among main chain atoms (N, C, Cα and O). Two atoms are in steric clash if their distance is smaller than the 70% of sum of the respective van der Waals radii.Ranking
The final step is the ranking of the remaining candidate loops. The ranking is based on a composite function that takes into account sequence and structural information in the form of main-chain phi/psi angle propensities. The sequence similarity score is calculate using the conformation similarity weight matrix described here The propensity score measures the compatibility of observed and expected dihedral angles of each residue of the candidate loop in the corresponding position of the query. Main chain conformation definitions and propensities are defined according to the p15 propensities table described here. Sequence and propensity scores are combined and normalized by computing Zscores.Submission form
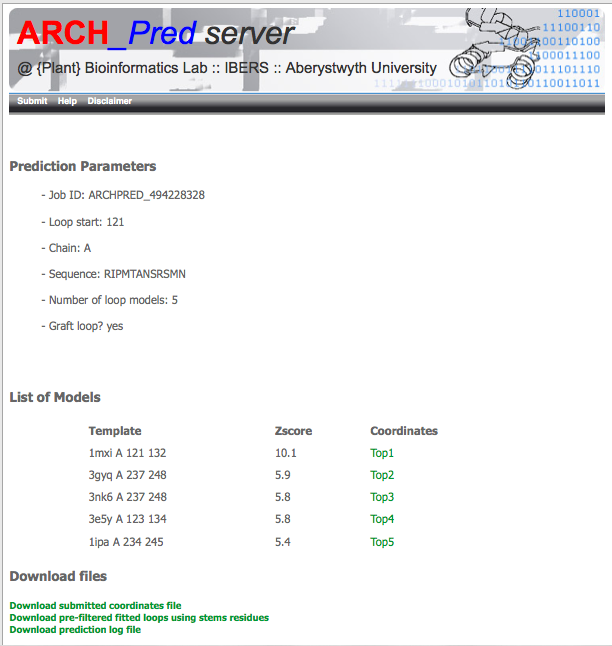
Prediction parameters
The first section of the submission form allow users to upload the coordinates of the protein structure containing the missing loop(s). The atomic coordinates must be on standard PDB format. Users have to define the starting residue of the missing loop and the chain ID.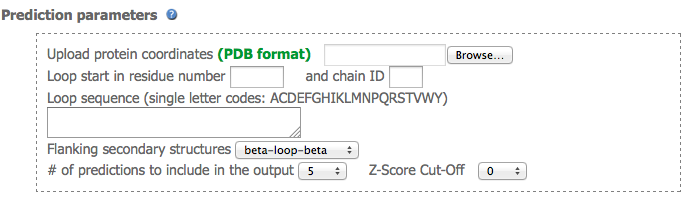
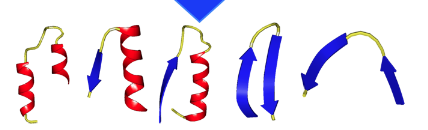
From left to rigth: alpha-loop-alpha; alpha-loop-beta; beta-loop-alpha; beta-loop-beta (hairpin); beta-loop-beta (link). Note that both hairpins and links are considered beta-loop-beta motifs.
Finally, users select the number of loop structure to be returned by the prediction (10 maximum) and the Z-score cut off.IMPORTANT The numbering of the uploaded coordinates must be consistent and in agreeement with the missing loop. For instance, if predicting a missing loop of length 14 and the loop stars at residue 17 and chain A, then the PDB file should look like this:
... ATOM 826 N PHE A 14 49.206 19.282 50.320 1.00 54.69 N ATOM 827 CA PHE A 14 48.606 19.107 51.603 1.00 55.79 C ATOM 828 C PHE A 14 47.836 17.809 51.723 1.00 56.22 C ATOM 829 O PHE A 14 48.078 17.051 52.638 1.00 55.30 O ATOM 830 CB PHE A 14 47.690 20.257 51.882 1.00 56.82 C ATOM 837 N LEU A 15 46.893 17.551 50.827 1.00 57.29 N ATOM 838 CA LEU A 15 46.182 16.280 50.875 1.00 59.26 C ATOM 839 C LEU A 15 47.047 15.013 50.740 1.00 59.98 C ATOM 840 O LEU A 15 46.727 13.971 51.280 1.00 60.70 O ATOM 845 N GLU A 16 48.121 15.088 49.997 1.00 60.70 N ATOM 846 CA GLU A 16 48.980 13.935 49.837 1.00 61.99 C ATOM 847 C GLU A 16 49.482 13.456 51.186 1.00 62.42 C ATOM 848 O GLU A 16 49.617 12.256 51.424 1.00 62.45 O ATOM 946 N VAL A 31 25.144 19.607 51.305 1.00 51.18 N ATOM 947 CA VAL A 31 25.010 20.708 52.246 1.00 52.08 C ATOM 948 C VAL A 31 23.603 20.835 52.761 1.00 52.94 C ATOM 949 O VAL A 31 22.705 21.156 52.037 1.00 54.54 O ATOM 953 N THR A 32 23.403 20.559 54.023 1.00 54.13 N ATOM 954 CA THR A 32 22.075 20.604 54.610 1.00 55.49 C ATOM 955 C THR A 32 21.767 21.988 55.149 1.00 56.13 C ATOM 956 O THR A 32 20.606 22.329 55.356 1.00 56.55 O ...If the numbering is not consistent, coordinates will be deleted when the PDB file is parsed
Post-prediction optimization
ARCH_Pred allow the post-prediction optimization of predicted loops. This includes the grafting of the loop in the the protein structure and the refinement using modeller. In this case, the server returns individual protein structures with grafted loops. If unchecked, the server will return the coordinates of the protein and the candidates loops in a NMR style PDB file where MODEL 1 is the frame protein and the candidate loops structures (main chain traces) MODEL 2 and above.
Results web page
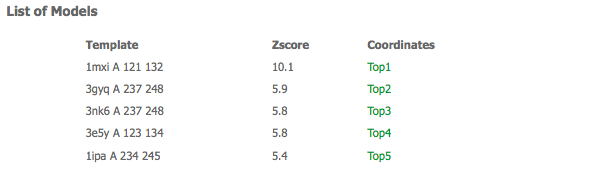
If the prediction is sucessful the web server will return an web page similar to the one shown below. Should an error occur during the prediction, the web page will show an specific error message and a link to the prediction log file available to download for inspection