Frag'R'Us
Frag'R'Us at a glance
Frag'R'Us is a web application to extract and characterize supersecondary, or smotifs, as defined in this publication, from protein structures. Each smotif is characterized by its geometry as defined by 4 internal variables: D (Ang), delta (or hoist angle), theta (or pack angle) and rho (or meridian angle). The figure below shows the schematic representation of the geometry descriptors. More information is available here and here.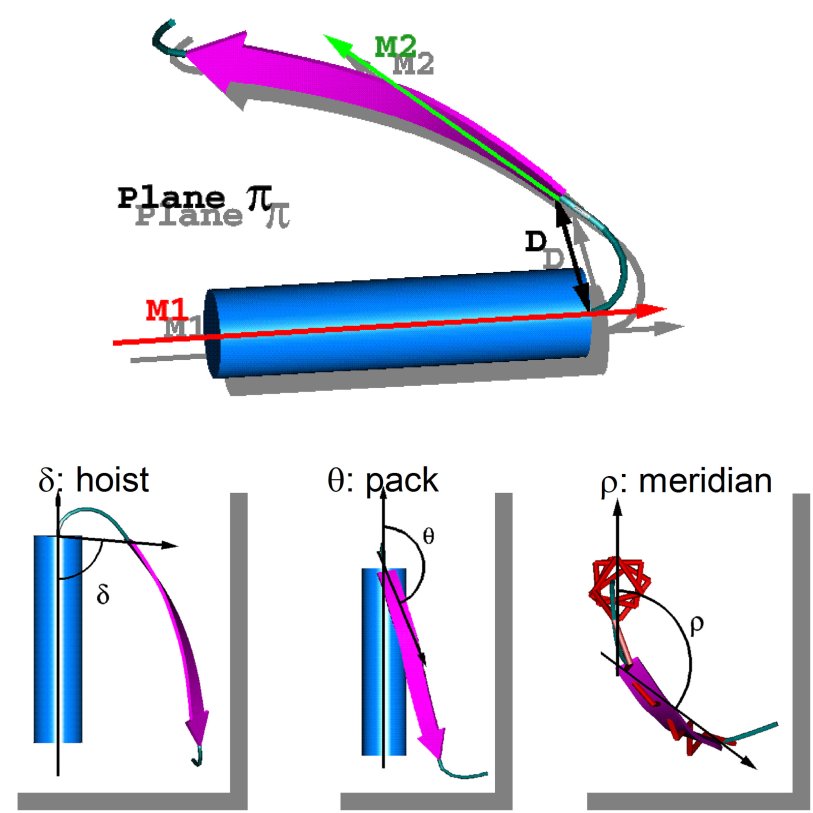
-
1. Extract smotifs and assign the geometry values: D, delta, theta and rho angles.
-
2. Perform a search on a library of smotis (40: non-redundant; 90: redundant) and
extract those smotifs that match the geometrical restraints of the query smotif. This
task have several application such as loop modeling or protein-design.
-
3. Performs the structural overlaid of smotifs obtained in 2 to the reference smotif used as a query.
Why using Frag'R'Us
Frag'R'Us provides alternative main-chain loop conformations between two flanking secondary structures by comparing the geometry of a query smotif and a large library of smotifs extracted from protein structures. Frag'R'Us can therefore be used in computational protein design, either in enzyme design by providing different conformation of catalytic loops or protein-protein or protein/DNA interface design by providing alternative conformations of interface loops. Frag'R'Us is also useful in loop structure prediction by providing alternative conformations of loops that are length independent. Note: if the sequence of the missing loop is know, then ArchPRED is a better tool. The modulaty of smotifs can be also applied in build-up approaches in protein structure prediction and the refinements of protein crystal structures.Submission form
Upload section
The first section of the submission form allow users to either select a PDB code or upload the coordinates of the protein of interest. The atomic coordinates must be on standard PDB format and the chain ID must be provided.
Extraction smotifs
The second section defines how the secondary structure will be assigned. Users can either choose DSSP or provide their own assignments. If DSSP is selected, then users should defined which DSSP will be used: an alphabet of 3 states implies that only those residues in an 'H' or 'E' states (as DSSP) are considered regular secondary structures, the rest of states I,T,S,C,B,G are assimilated to loop conformation. An alphabet of 5 implies that E and B states are considered betas, and H and G states helices. If the secondary structure of the protein is provided, then only H:helice, E:strand and C: rest coding must be use. Users should also define the minimum size for a beta strand and alpha helix. Values are given by default and perhaps those are a good starting choice.
Search smotifs
The third section defines the search parameters. Database: 40 and 90 are a non-redundant and redundant library of smotifs respectively. The maximum difference in geometry values between query and target smotifs is also defined in this section. For instance, a maximum variation of D, delta, theta, and rho values of: 0.5, 5, 5, 10 respectively implies that smotif(i), smotif(j) would have the same geometry if |(D,delta,theta,rho)(i) - (D,delta,theta,rho)(j)| <= (0.5,5,5,10). User can restrict the search of smotifs to those that have a given number of residues in the loop residues. By default the search is length-independent. This parameter is useful if looking for loops of a defined length matching a given geometry.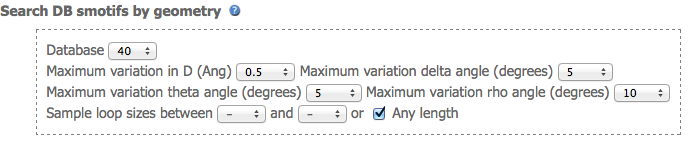
Tasks
The final section defines the task to perform. 1. Simply extract smotifs and assign geometry. 2. Perform also a search of DB based on geometry, and 3. Include the structural superposition of smotifs to the query. If task 3 is selected, i.e. structural fitting of candidate and query smotifs, then users can adjust the parametres of the steric fitting activating a filter to eliminate smotifs that clash with the frame proteins and the RMSD of stems residues; 2 Ang by default.
Results web page
Upon completion of the job, the server generates a web page with all the information in differents selections. In the first selection it can be found the parameters selected for the job. This include the structure submitted to the server, the chain ID, and geometry features among others. Below the parameters selection there is the sequence of the protein that will be used to locate the different smotifs upon selection, see below.
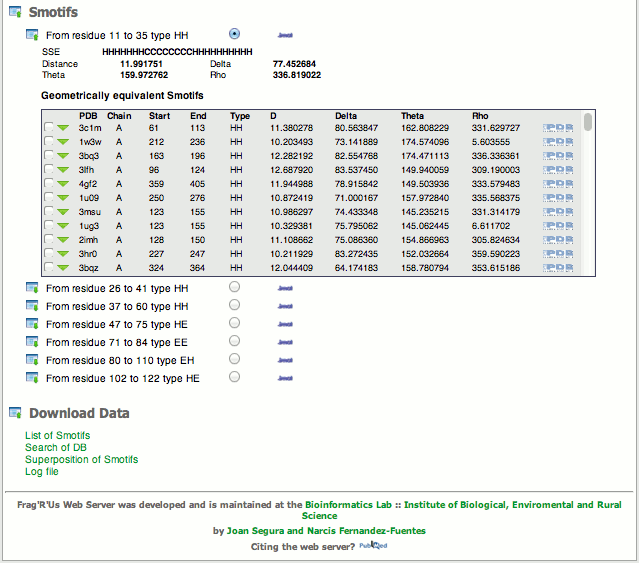
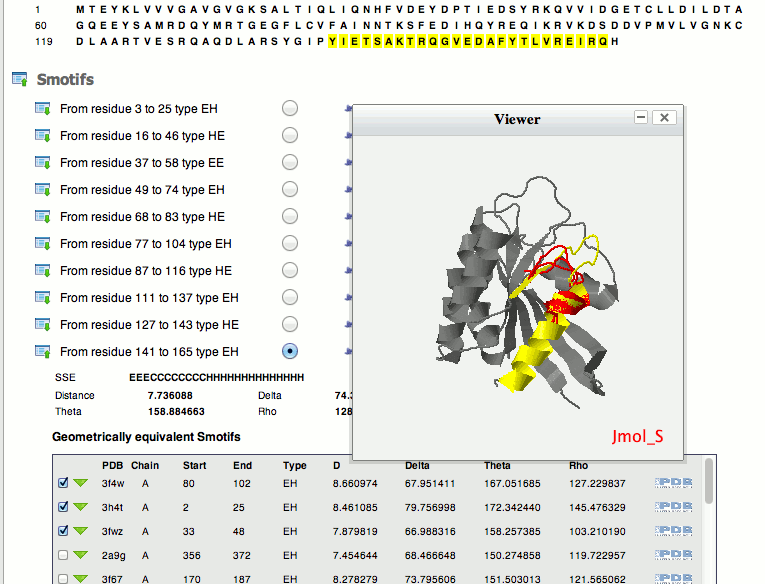
-
1. geom.out It is a tab-delimited text file with the list of smotifs extracted from the protein structure.
The contents of column 1 to 12 are: Pdb file, Chain ID, Smotif Type, Start residues (AS IN PDB FILE), Loop size, Nt
secondary structure size, Ct secondary structure size, Sequence, Secondary Structure (as in DSSP), encoded Phi/Psi string angles, D (Ang),
Delta (degrees), Theta (degrees), Rho (degrees). This is the output file is task 1 was selected.

-
2. search.out It is a tab-delimited text file. A MOTIF : tag highligths the query smotif and below there is a list of smotifs extracted
from the library that match geometrical restraints. This is file and the previous one are the output files is task 2 was selected.

-
3. super.tar.bz2 It is a compressed tar file containg atomic coordinates of smotifs extracted from the library and structurally superposed to the query smotifs.
The format of the atomic coordinates is the standard PDB file used for NMR structures, i.e. each of the smotifs superposed to the query smotifs are delimited by 'MODEL'
and 'ENDMDL' tags; MODEL 1 is always the query smotif. The header of the file contains information regarding
RMSD values and transformation matrices among other useful information. An example showing a full search using a protein structure is shown below.
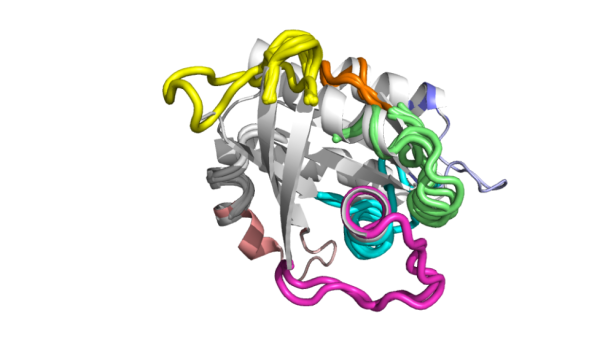
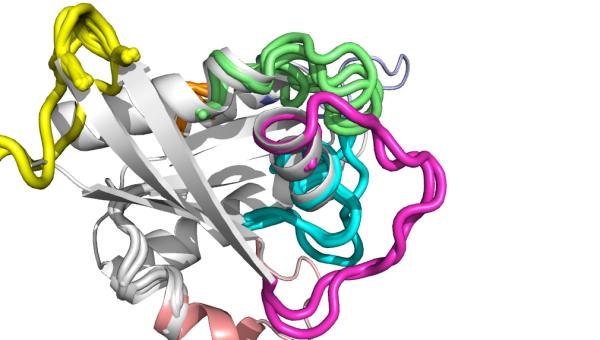
No results?
Any errors incurred during the submission process will be reported prior to the submission of the job. These include:- Error in the formatting of the coordinates files, should be standard PDB format.
- Error in PDB code if not present in our local database.
- Missing information in the submission form: missing chain ID; method to calculated the secondary structure: i.e. DSSP or own assigment.
- Different size PDB file and secondary structure if provided by user. The number of C-alphas in the PDB file are checked against the lenght of the secondary structure string and an error is reported if different.
- Task 1. The protein does not contain smotifs. The minimum unit for Frag'r'Us to work is a regular secondary structure (alpha/beta-strand) followed by a loop and by a second regular secondary structure If this minimum unit is not present, then Frag'r'Us will fail to located smotifs.
- Task 2. The search does not yield any results. If parameters for the search of suitable smotifs is too restrictive, i.e. tolerance of D, delta, theta and rho variable is set too low, there is the risk that no geometrically equivalent smotifs will be found. If this happens, increase the tolerance values. The default values are a good start.
- Task 3. The fitting of smotifs does not yields any results. This could be due, if clashes filtes is active, to the fact that smotifs does not fit the new protein without having serious steric clashes. To overcome this situation, a new search could be done using more tolerance geometry matches, that will increase the number of candidate smotifs.
- Protein chain breaks, missformating coordinate files, etc. Errors in the coordinates file will affect the process, so it is advisable to check the numbering and consistency of the PDB file.